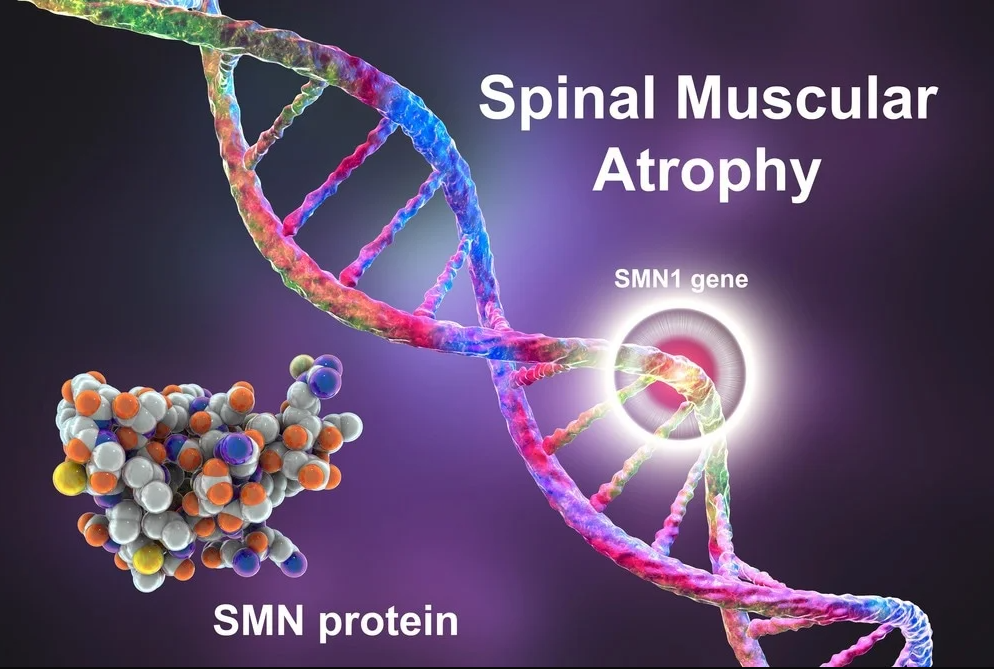
Spinal Muscular Atrophy (SMA) is a genetic condition that primarily impacts the motor neurons of the spinal cord. These neurons are vital for muscle activities such as crawling, walking, neck control, and even swallowing and breathing. SMA is characterized by a loss of motor neurons, which leads to muscle weakness and atrophy, or the wasting away of muscles.
What is Spinal Muscular Atrophy?
Spinal Muscular Atrophy affects approximately 1 in 10,000 babies, making it one of the more common rare diseases. Despite its rarity, the impact it has on the affected individuals and their families is significant. The disease can present at various stages of life, from infancy to adulthood, with varying degrees of severity. The most severe form is Type 1 SMA, also known as Werdnig-Hoffmann disease, which often presents in the first months of life and is typically fatal without intervention.
The root cause of SMA is a deficiency in a protein known as survival motor neuron (SMN) protein. This protein is crucial for the maintenance and survival of motor neurons. Without sufficient SMN protein, these neurons gradually degenerate, leading to muscle weakness and atrophy.
SMA Types Explained
Traditionally SMA symptoms are divided into four types depending on the age at which symptoms appear and the related severity of muscle weakness, based on the motor milestones achieved. Even within these types there is a wide spectrum of severity as SMA affects everyone differently.
Babies diagnosed with SMA type 1 show symptoms within the first weeks or months of life (before six months of age) and they do not achieve the ability to sit independently. Babies with SMA type 1 have generalised muscle weakness and limited movement. They typically have a weak cry and difficulty coughing due to weak breathing muscles. These babies experience problems with swallowing and suffer from frequent respiratory infections. Life expectancy for babies with SMA type 1 is very limited but can vary. In many cases, babies may live for less than two years; especially without ongoing respiratory intervention. However, with round-the-clock care and respiratory support, some children with SMA type 1 may survive into their teens or longer. Type 1 is the most common type of SMA. Around 60% of cases of SMA are classified as type 1.
Symptoms of SMA type 2 appear between seven months and 18 months of age. Children are diagnosed with SMA type 2 if they were strong enough to maintain sitting position at some point, and some may have achieved the ability to stand, but they never gain the ability to walk unaided. Just like in SMA type 1, children and adults with SMA type 2 often have difficulties swallowing and are prone to respiratory infections due to weakness of the muscles used for breathing. They all also require mobility aids (for example, a wheelchair) due to progressive muscle weakness. However, with good care, they live to become teenagers and adults. Stronger people with SMA 2 may work, some start families.

When the first symptoms appear after the person learned to walk independently, the disease is classified as SMA type 3. People with this type still experience significant muscle weakeness over time and the vast majority soon lose the ability to walk or stand, but they tend to live full lives and oftentimes achieve a lot in personal and professional life.
Very rare cases when SMA symptoms develop in an adult person are termed as SMA type 4.
All the four SMA types have the same genetic cause and all drugs that act on SMA-causing genes should be of help in all types of the disease, although perhaps to a varying degree. However, we believe, along with many healthcare professionals, that all people with SMA will be helped greatly if treated with any of the upcoming powerful drugs for SMA.
For more information see NHS SMA Symptoms.
The Genetics Behind Spinal Muscular Atrophy
SMA is inherited in an autosomal recessive pattern. This means that an individual must inherit two mutated copies of the gene associated with the condition – one from each parent – to be affected by SMA. The parents, each carrying one mutated copy, are known as carriers. They typically do not show signs or symptoms of the condition.
The gene in question here is the SMN1 gene, which provides the instruction for producing the SMN protein. In most people, this gene exists in multiple copies. However, in SMA, a mutation results in the deletion or alteration of this gene, rendering it unable to produce functional SMN protein.

Humans also have a similar gene called SMN2, which can produce the SMN protein but in far lesser amounts. The interesting point is the variability in SMN2 copies among individuals. Those with more copies of the SMN2 gene tend to have milder forms of SMA, as the SMN2 gene partially compensates for the loss of SMN1.
Testing and Treatment
Today, newborn screening for SMA is available and recommended in many countries. Early detection of SMA can significantly impact the quality of life of affected individuals, as it allows for early intervention. Genetic testing can also identify carriers of SMA mutations, providing vital information for family planning.
It is estimated that one in 40 adults carry the faulty SMN gene, which is why newborn screening is absolutely critical. Not only from a family planning perspective but the sooner treatment is started the better for the afflicted. At present the UK does not provide newborn screening for SMA and this is something that we are working hard to try and resolve.
Advancements in medical science have made it possible to manage SMA better than ever before. Treatments including gene therapies, SMN protein enhancers, and neuronal modulators have shown efficacy in managing this condition. These therapies work to increase the production of SMN protein or slow the progression of the disease.
In conclusion, while SMA is a severe genetic condition, understanding its genetic underpinnings has led to significant strides in managing the disease. Genetic testing and early intervention can make a significant difference in the lives of affected individuals. Furthermore, continuous research into SMA offers hope for even more effective treatments in the future.
Unveiling the Complexities of SMA Genetics
While the basic genetic inheritance pattern of SMA is fairly well understood, the situation gets slightly more complicated when you consider the variable nature of the SMN2 gene. As we’ve mentioned earlier, SMN2 is a “backup” gene that can produce the SMN protein, albeit less efficiently than SMN1.
But the amount of functional SMN protein that SMN2 can produce varies from person to person, and this is largely due to the number of SMN2 gene copies an individual has. Typically, an individual with SMA can have anywhere from one to eight copies of the SMN2 gene. More copies generally equate to a milder form of the disease.
For example, a child with SMA Type 1 might have one or two copies of the SMN2 gene, while a child with a milder form, SMA Type 3, might have three or four copies. Researchers use this information to predict disease severity and guide treatment options, illustrating the remarkable significance of SMN2 in managing SMA.
Unraveling the Future of SMA
As research continues, there’s ongoing hope for advancements in SMA treatment. A key area of research is aimed at enhancing the function of the SMN2 gene to produce more SMN protein. This approach could provide a more definitive way to manage SMA, mitigating symptoms and possibly even reversing motor neuron damage.
Another promising area of research is in the field of stem cell therapy, which involves introducing healthy cells to replace the degenerating motor neurons. While this research is still in its early stages, the potential for radical treatment improvements is substantial.
The Role of Genetic Counseling
For families affected by SMA or those identified as carriers through genetic testing, genetic counseling plays a crucial role. Genetic counselors can help interpret complex genetic information and guide individuals or couples through their reproductive choices.
Prenatal testing or preimplantation genetic diagnosis (PGD) during IVF can identify embryos affected by SMA. Such technologies offer couples the choice to avoid passing the condition onto their children. The decisions surrounding these options are profoundly personal and can be emotionally charged. Genetic counselors provide valuable support and guidance throughout this process.
Concluding Thoughts
SMA is a complex genetic disorder that poses significant challenges to those affected and their families. But it’s also a condition where we’ve seen substantial scientific advancements, particularly in the understanding of its genetic foundations. This knowledge has fueled the development of treatments that, while not yet a cure, are drastically improving the lives of those with SMA.
Despite the complexities and challenges, the future for individuals with SMA is brighter than it’s ever been. As researchers continue to delve into the intricacies of the SMN1 and SMN2 genes, and as treatments continue to advance, there is every reason to hope for a future where SMA can be effectively managed or perhaps even cured.

